FastPCR
- Java application is based on FastPCR software for Windows and provides comprehensive and professional facilities for designing primers for most PCR applications and their combinations: standard, multiplex, long distance, inverse, real-time, Xtreme Chain Reaction (XCR®), group-specific (universal primers for phylogenetically related DNA sequences) or unique (specific primers for each from phylogenetically related DNA sequences), bisulfite PCR primer design, polymerase extension PCR multi-fragments assembly cloning (OE-PCR), Loop-mediated Isothermal Amplification (LAMP); microarray design; Allele-Specific PCR (AS-PCR) - KASP™ (Kompetitive Allele Specific PCR) or PACE™ (PCR Allele Competitive Extension) based genotyping assay design for biallelic discrimination of single nucleotide polymorphisms (SNPs) and insertions and deletions (Indels) at specific loci;
- in silico (virtual) PCR for primers and probes searching or in silico PCR against genome(s) (or a list of chromosome) - prediction of probable PCR products and search of potential mismatching location of the specified primers or probes;
- comprehensive primer test, the melting temperature calculation for standard and degenerate oligonucleotides including LNA and other modifications, primer's PCR efficiency and linguistic complexity, and dilution and resuspension calculator;
- analyzes different features of multiple primers simultaneously, the melting temperature, GC content, sequence linguistic complexity, primer PCR efficiency and molecular weight, the extinction coefficient, the optical density (OD);
- primers (probes) are analyzed for all primer secondary structures including the alternative hydrogen bonding to Watson-Crick base pairing such as G-quadruplexes or wobble base pairs (like G-G, G-T, G-A), self-dimers and cross-dimers in primer pairs;
- tool calculate Tm for primer dimers with mismatches for pure and mixed bases using averaged nearest neighbour thermodynamic parameters and for modifications (inosine, uridine, or LNA);
- tool for identifying simple sequence repeat (SSR) loci by analysing the low complexity regions of input sequences;
- tool for restriction I-II-III types enzymes analysis, find or create restriction enzyme recognition sites for coding sequences;
- tool for searching for similar sequences (or primers);
- translates nucleotide (DNA/RNA) sequences to the corresponding peptide sequence in all six frames for standard and degenerate DNA and modifications (inosine, uridine);
- Polymerase Chain Assembly (PCA) - created to automate the design of oligonucleotide sets for long sequence assembly by PCR;
- the program includes various bioinformatics tools for patterns analysis of sequences with GC:(G-C)/(G+C), AT:(A-T)/(A+T), SW:(S-W)/(S+W), MK:(M-K)/(M+K), purine-pyrimidine (R-Y)/(R+Y) skews, CG% and GA% content and the melting temperature and considers linguistic sequence complexity profiles;
- Efficient and complete detection of different types of repeats and any type of tandem repeats, from short tandem repeats (microsatellites) to large satellite sequences with a summary table of the types and locations of the found repeats.
System requirements
The tools are written in Java and require the Java 24 Runtime Environment (Java SE Platform).
 |  |
Comparison of primer design and oligonucleotide analysing tools versus other most popular on-line primer design and analysing packages
The program structure
FastPCR Manual
Two independent text editors at different TAB: Sequences, Pre-designed primers (probes) list.
TAB: Sequences - for all sequences that will be analyzed, it can be a list of primers or sequences of chromosomes.
TAB: Pre-designed primers (probes) list - only for a list of primers, which will be analyzed in silico PCR, and for PCR to check the compatibility of these primers with primers that will be designed.
Import sequences from a clipboard or right-click mouse displays a contextual menu or from File.
Import sequence(s), about sequence formats
The popup menu:

 Open file(s) into current TAB Ctrl-O - open any text, .HTML and .RTF file with DNA sequence(s), or .XLS TAB-column formatted text files)
Open file(s) into current TAB Ctrl-O - open any text, .HTML and .RTF file with DNA sequence(s), or .XLS TAB-column formatted text files)
 Load file(s) from the folder Ctrl-G directly into memory without opening files into editor; small or large text file with DNA sequence(s) at FASTA format or TAB/Space-column formatted text files
Load file(s) from the folder Ctrl-G directly into memory without opening files into editor; small or large text file with DNA sequence(s) at FASTA format or TAB/Space-column formatted text files
 Direct analysis of all file(s) Ctrl-J from the folder without opening files into editor; small or large text file with DNA sequence(s) at FASTA format or TAB/Space-column formatted text files
Direct analysis of all file(s) Ctrl-J from the folder without opening files into editor; small or large text file with DNA sequence(s) at FASTA format or TAB/Space-column formatted text files
Importing sequences must be at the same format.
Prepare your sequence data file using a text editor (Notepad, WordPad, Word), and save in ASCII text format (plain text) or Rich Text Format (.RTF).
You can type in "Sequence editors" or import nucleotide sequence(s) from file or from the clipboard (Shift-Insert, Ctrl-V) as simple text or Excel sheet or Word table (two columns), the table with TAB or whitespace separators.
For individual, selective options and task, sequences need convert to FASTA format with “>”, these options have a highest priority. Any of these following commands must be written AFTER the sequence name or “> ” (these commands are not case sensitive) and press Enter and the end of line. The commands can occupy any place in the command line.
When sequences are imported you may edit the sequences in general or additional editors and immediately visualize the result of editing. Press Ctrl-R switch on-typing sequence interpretation to on or off. You can modify a nucleotide sequences by inserting, deleting and replacing sequence fragments.
Degenerate primer sequences are accepted:
IUPAC DNA degenerate code is an extended vocabulary of 15 letters which allows the description of ambiguous DNA code. Each letter represents a combination of one or several nucleotides:
B=(C,G,T), D=(A,G,T), H=(A,C,T), K=(G,T), M=(A,C), N=(A,C,G,T), R=(A,G), S=(G,C), V=(A,C,G), W=(A,T), Y=(C,T). U=Uracil; I=Inosine; and LNA: E=dA, F=dC, J=dG, L=dT.
Raw format (ASCII)
Like a text/plain format without white space and TABs. It read only standard IUB/IUPAC amino acid or nucleic acid codes characters and rejects anything else, low- and upper-case insensitive. Digits or else are removed and ignored (but Tab and space characters with combination end line character (Enter press) can be interpreted as column format). Here are some examples of raw formatted sequence:ataaattcttattttgacactcaccaaaatagtcacctggaaaacccgctttttgtgaca
 Press at tool bar for converting sequences into FASTA format (Alt-1), for checking correct format reading.
Press at tool bar for converting sequences into FASTA format (Alt-1), for checking correct format reading.  Reverse-Complement transformation (e.g. 5'-acacacc ⇒ 5'-ggtgtgt) (Alt-2).
Reverse-Complement transformation (e.g. 5'-acacacc ⇒ 5'-ggtgtgt) (Alt-2). Reverse (e.g. 5'-acacacc ⇒ 5'-ccacaca) (Alt-3).
Reverse (e.g. 5'-acacacc ⇒ 5'-ccacaca) (Alt-3). Antisense (e.g. 5'-acacacc ⇒ 5'-tgtgtgg) (Alt-4).
Antisense (e.g. 5'-acacacc ⇒ 5'-tgtgtgg) (Alt-4).
FASTA format description
FASTA format have a highest priority and is simple as the raw sequence proceeded by definition line. The definition line begins with a “>” sign and optionally followed immediately by a name for the sequence with using any length and amount of words. Many sequences can be listed in the file, the format indicating a new sequence at each new “>” symbol found. It is important to press Enter at the end of each line after “>” to help FastPCR recognize the end and beginning of sequence and sequence’s name. Make sure the first line starts with a “>” and has (has not) a header description.
The description must be contained within one line and not run into 2 or more lines. The sequence starts directly on next line. As for the previous raw data format, sequences must be in the standard IUB/IUPAC amino acid or nucleic acid codes, any other characters - digits, spaces, TAB characters or else are ignored, low- and upper-case insensitive:>
cggccgagatcaggcgatgcatg>
acgacgacgcagctatattacag
Tables format description
You can directly import the table from text file or from the clipboard via copy and paste operations from Microsoft Word or Excel sheet (or OpenOffice), or primer’s list from FastPCR's", or the table with TAB or whitespace separators. Software reads only first two columns with names and sequences, or first three/four columns for primers and probe:
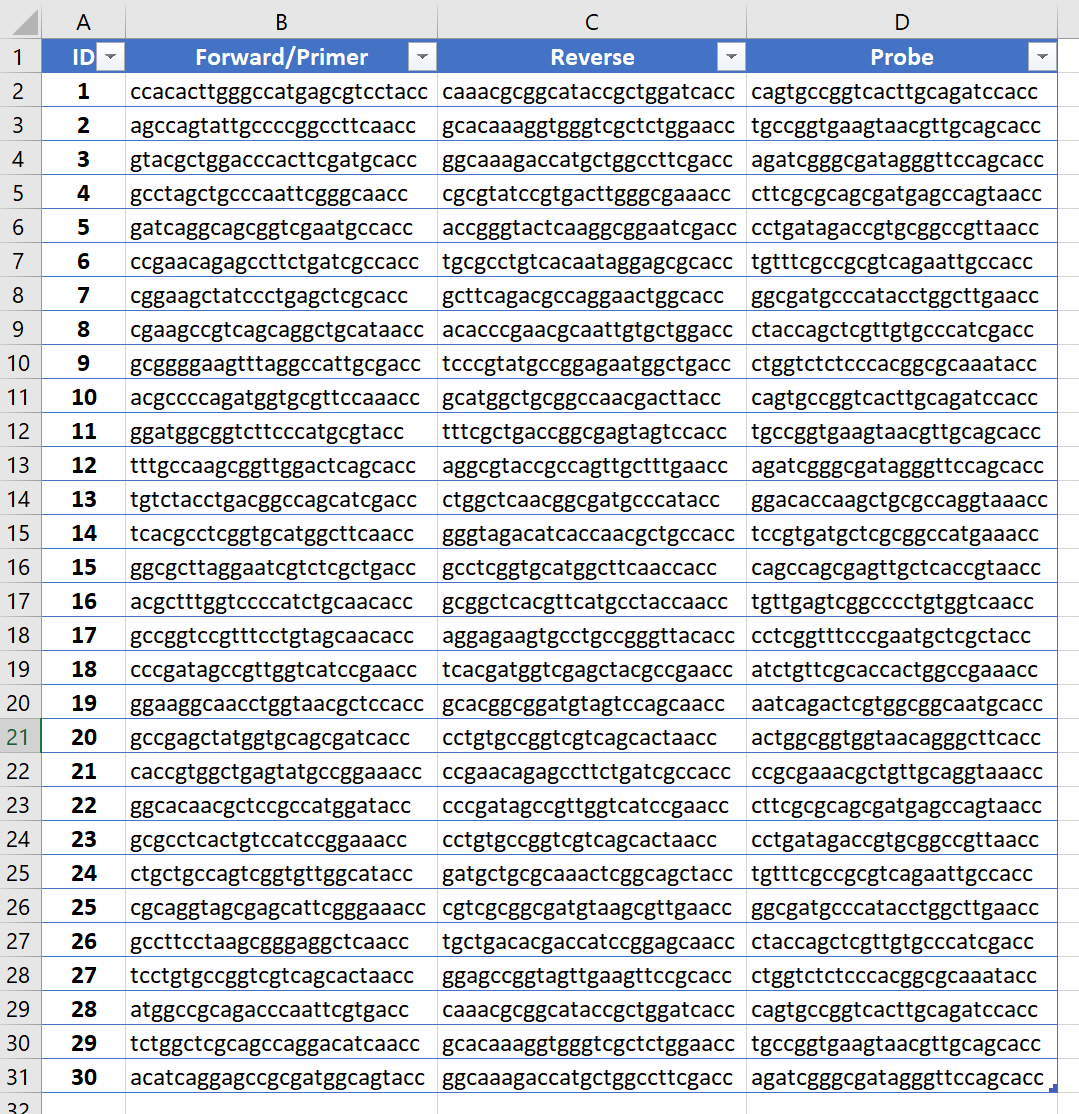
To check the correct format was read, look at the information in Sequence TAB and under text editor in the status bar:

As for the previous raw data format, sequences must be in the standard IUB/IUPAC amino acid or nucleic acid codes, any other characters - digits, spaces or else are ignored, low- and upper-case insensitive. Tab character or spaces are used for recognition columns. Other simple table format is with or without name for primers (probes or else); name is replaced by single space (space inside sequence not allowed) and the end of each sequence, press Enter is necessary:
- acgaatcgtattcaagcctgc
- gcgtcatctggctgctacctcga
- cgagcttagtcttcaacgccaa
- agaggacgctcgtgtctttcggac
- gctcacgtcaaagtcttgtccgag
In case using sequence’s name, no space inside names and sequences are allowed.
Software always indexing each sequence from 1 to N, therefore doesn’t matter if some sequence’s name are the same or absent:
1 acg aat cgt att caa gcc tgc
ccg tca tct ggc tgc tac ctc ga
cga gct agt ctt caa cgc caa
1 aga gga cgc tcg tgt ctt tcg gacGeneral PCR primer options
For PCR primers are usually 18-35 bases in length and should be designed such that they have complete sequence identity to the desired target fragment to be amplified. Parameters controllable by the user are primer length (12-500nt), melting temperature calculated by nearest neighbour thermodynamic parameters, theoretical primer PCR efficiency (quality at %) value, primer CG content, 3’end terminal enforcement, preferable 3’termini nucleotide sequence composition in degenerated formula and additional sequence at 5’ termini.
The other main parameters used for primer selection are: the general nucleotide structure of the primer such as linguistic complexity (nucleotide arrangement and composition); specificity; the melting temperature of the whole primer and the melting temperature at the 3’ and 5’ termini; a self-complementarity test; secondary (non-specific) binding search:

- Quality limit (PCR primer amplification efficiency), this abstract value of the ‘primer quality’ describes thus the level of primer/PCR successfulness; this value varies from 100% for the “perfect or ideal” to 0% for the "worst" primer. A “perfect” primer has a wider range of executable temperatures. A program is select the best primer with optimal range of executable temperature, which allowed to design qualified primers (probe) for any target sequences with any CG and repeat contents.
- Linguistic complexity control: the complexity values were converted to a percentage value, in which 100% means maximal ‘vocabulary richness’ of a sequence.
- 3'-End composition (5'-3'): you can choose with 5’ or 3’ ends is most preferable for all primers or probe. Minimal size is 2 letter (maximum – primer length), “5’-nn-3’” is any, “5’-sww-3’” – all variants for 5’-(C/G)(A/T)(A/T)-3’”. You can type one or more variants with space between them and with the same length.
- С >> T bisulfite conversion (bisulfite modified genome)
Sequence, design of specific PCR primers for in silico bisulphite conversion for both strands - only cytosines not followed by guanidine (CpG methylation) will be replaced by thymines:
5’aaCGaagtCCCCa-3' 5’aaCGaagtTTTTa-3'
||||||||||||| -> ||||||:|::::|
3’ttGCttCaggggt-5' 3’ttGCttTaggggt - Alternative amplification (non-specific binding ) control: the program will analyse primers at current sequence and at "Reference dataset (non-specific primer binding test)" (using quick hash-table alignment) and look for the presence of possible additional complement sites for each primer (probe), which would result in non-specific PCR product. Usually, the site-specificity of the primer can be checked by performing a sequence homology search (e.g. blastn) through all known template sequences in the public genome database such as National Center for Biotechnology Information (NCBI), but it is no necessity, our experience, the almost all problems can be simply solving using any the BAC sequence(s) (about 100,000 or more bases) from analysing genome, because only high copy number repeats will compromise the performance of unique PCRs so, unless the experiments are planned specifically to exploit them, they are best avoided.
PCR primers and probes design command lines
Any of these following commands must be written AFTER the sequence name or “>” (these commands are not case sensitive) and press Enter and the end of line. The commands can occupy any place in the command line.
PCR set-up examples
PCR Output Result
Application automatically generate results in "Results Tab" at tabulated format (ready for transferring to Excel sheet via copy and paste operations); the output results is easy to save as .XLS Text file, compatible for Excel or Open Office: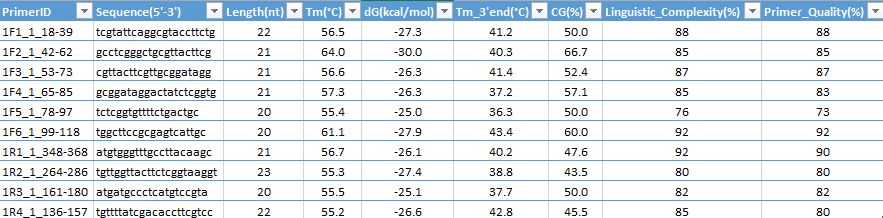
Primers ID (Identifiers) designed format:

PCR product output, each line contains compatible primer pair (Forward and Reverse primers):
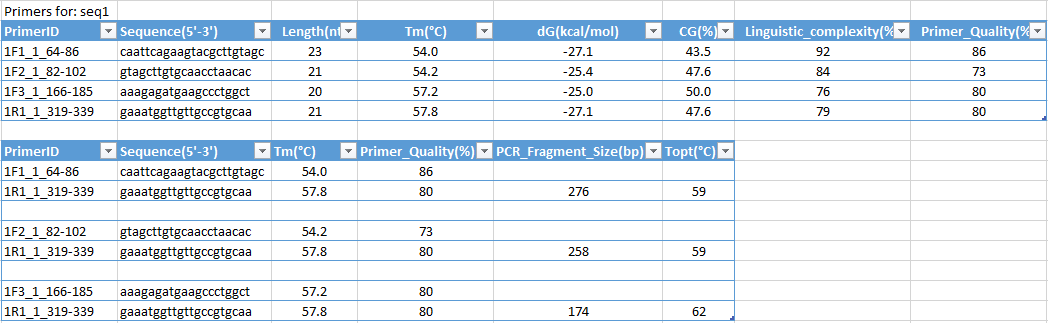
Multiplex primer design
The user can also input options for the PCR product involving the minimum product size differences among the set of designed primer pairs. It also allows to set primer design conditions individually for each given sequences or using common options. The individual setting have highest priority to PCR primer or probe design than general settings. The result includes primer sequences for individual sequences, their compatible primer pairs with product size and annealing temperature and final result for compatible primer pairs for each sequence with all information includes primer pair sequences, product size and annealing temperature. It is ideally to design all primer pairs with near equal annealing temperature in single reaction. For most cases the multiplex PCR conditions are resisting to a small variation (up 7°C) of Ta between all primer pairs and PCR products. Synchronizing Tm for primer pair user can control from “Primer Design Options” or with command: -ptms5.
The annealing temperature must be optimal in order to guarantee effective amplification of the targets genomic sequence while minimizing the risk of unspecific amplification. To amplify the target genomic sequence effectively, the primers “quality” and properties should be highest. PCR primer design for multiplex PCR can be performed for standard or inverted PCR pairs or both of them. A minimum of two sequences must be implemented for this analysis. The program will find the compatible primer pairs for each sequence and will make a continuous numbering of pairs for all investigated sequences.
Another feature of the program, user can select not only compatible pairs of primers, as well as compatible single primers for different targets or sequences. That is, program can design both pairs of primers and single primers or only single primers for different targets:

Polymerase extension PCR for fragment assembly
Sequence-independent cloning, including ligation-independent cloning (LIC) requires generation of complementary single-stranded overhangs in both the vector and insertion fragments. Similarly, multiple fragments can be joined or concatenated in an ordered manner using overlapping primers in PCR.Annealing of the complementary regions between different targets in the primer overlaps allows the polymerase to synthesise a contiguous fragment containing the target sequences during thermal cycling, a process called 'overlap extension PCR' (OE-PCR). The efficiency depends on the Tm and on the length and uniqueness of the overlap. To achieve this, the programme designs compatible forward and reverse primers at the ends of each fragment, and then extends the 5' end of primers using sequences from the primers of the fragment that will be adjacent in the final product. The programme selects the overlapping area so that the primers from overlapping fragments are similar in size and in their optimal annealing temperature. The programme adds the required bases so that the Tm of the overlap is similar to or higher than the Tm of the initial primers. Primers are tested for dimers within the appropriate primer pair.
Group-specific (family-specific primer set) and unique PCR primers design
The group-specific amplification also call as family specific or universal amplification is most important tool for comparative studies of genes and genomes, including studies of evolution and cloning new sequences. The specific sequences that link to concrete organism can be discovered by DNA polymorphism in these conservative genome regions (genes, transposable or repeat elements). For detection DNA polymorphism in relative sequences will help with design PCR primers around this polymorphic region.
The overall strategy of designing group-specific PCR primers is standard PCR design of only to regions of sequence common to all sequences (hash-table base alignment).
The test primer complementarity performed with fast no gap local hash-table alignment includes parameters for amount of mismatches at the 3’-end of primers and primers similarity to target sequence.
Program automatically specify the alignment parameters (the same as for in silico PCR) for primers searching – “initial searching word size, >3 (default = 7), nt”, important length of 3'-end, 5...20, nt for testing mismatches, minimal complement primer length (>12, nt) and the local similarity (default = 80%)”.
An output PCR result contains the group-specific PCR primers from each sequence and compatible primers combination with product size and temperature annealing.
FastPCR automatically designs larger sets of universal primer pairs for all given related sequences, identifies conservative regions without sequence alignment and generates suitable primers for all given sequences. All steps of algorithm are automatic and you can influence to the general options for primer design and alignment options. FastPCR will work only with any source of related sequences as long as it is possible to found short consensus sequences. The quality of primer design is dependent on both on sequence relationship, phylogenetic similarity and suitability of the consensus sequence to the design of any good primers. Software is able to generate group-specific primers for each sequence independently, that suit for all sequences.
Unique PCR
The strategy for a unique PCR primer design is opposite to the group-specific PCR primers (probes) design. This case program search unique regions within a DNA sequence and automatically designing primers with minimal user intervention and maximum flexibility.
Calculation of optimal annealing temperature
The optimal annealing temperature (Ta) is the range of temperatures where efficiency of PCR amplification is maximal without non-specific products.
The most important values for estimating the Ta is the primer quality, the Tm of the primers and the length of PCR fragment.
Primers with high Tm´s (>60°C) can be used in PCRs with a wide Ta range compared to primers with low Tm´s (<50°C). The optimal annealing temperature for PCR is calculated directly as the value for the primer with the lowest Tm (Tmmin). However, PCR can work in temperatures up to 10 degrees higher than the Tm of the primer to favour primer target duplex formation, our empirical formulae:
 ,
,
where L is length of PCR fragment.
In our experience, almost all high-quality primers designed by FastPCR in the default or «best» («Long distance» or «Quantitative» PCR) mode provide amplification at annealing temperatures from 68 to 72°C without loss of PCR efficiency, and show good amplification in varying PCR annealing temperatures and when using different DNA polymerases and buffers.
In silico (virtual) PCR or primers (probes) searching
This in silico tool is very attractive for quick analysing primer or probe through target sequences, for determination primer (probe) location, orientation, efficiency of binding, complementarity and Tm calculation.
The prediction appropriated short or long primer (probe) annealing site is only one way for PCR product prediction. Primer can bind many predicted sequences in template(s), but only sequences with few mismatches (1 or 2 depends from place and nucleotide) at 3’end of primer can be used for polymerase extension. The last 10-12 bases at 3’end of primer are sensitive to initiation of polymerase extension and general primer stability on binding template site. Single mismatch at these last 10 bases at 3’end of primer depends from the position and the structure can slightly reduced the primer binding and PCR efficiency. This software allows simultaneously testing single primer or list of the individual primer or probe with any length thorough multiplex target sequences. This test control by primer complementarity to target sequence performed with fast no gap alignment.

The in silico PCR program is initiated by clicking on ribbon “In silico PCR”. The required input items can be grouped into three parts.
- The entire target sequences should be pasted in the “Sequences” TAB text area. Target sequences can be either a single file contains multiple DNA sequences or all files from the folder. For In silico PCR against whole genome(s) or a list of chromosomes, user must specify a directory for input. The program will be consistent, a file-by-file, look at each file to the DNA sequence position of the primers. The program analyses every single file by searching binding site or sites of the analysed primer.
- Single or primer list sequences to be tested: user can type manually or paste pre-existing primer’s list into the second TAB of “Additional sequence(s) or pre-designed primers (probes) list” into text editor. The amount of pre-existing primers is not limited on the primer pair.
- The searching parameters into the TAB of “Parameters for PCR Product Analysis” contain:
The boxes of “Minimal PCR Product length (bp)” and “Maximal PCR Product length (bp)” – has the default value between 50 and 10000 bp, allowing user to define the minimal and maximal size of the expected PCR product. Any amplicons larger than a defined value will be filtered out.
- “PCR product prediction” – is the default value to predict PCR product size and to search primer binding sites. The disabled option can be set to keep a user from changing the value until some other conditions have been met.
- “Circular sequence” – if circular DNA molecules, e.g. plasmid, mitochondria or plastids DNA etc. have to be analysed, the primers can produce one or two amplicons.
- “Restrict analysis for F/R primer pairs” – this option is used for analysis of primers list, where each pair of primers is united by the common name. Similar analysis can be carried out for primers with same names or names, which differ at the last letter – F/R. The program will recognize paired primers (Forward as F and Reverse as R). For this analysis, primers from the same pair(s) must have identical names and ending with “R or F” (e.g. “seq1R” and “seq1F” form a pair). The name length and structure (including "F" and "R" inside names) are not important. The program is not limited to one unique pair per primers: there could be several "Reverse" for one "Forward" primer. The searching of potential amplicons will be carried out only for these primer pairs, while other primers from list will be ignored.
Additional configuration settings allow to optimize the primer or probe binding site searching and increase the representativeness of the results. This is mainly determined by the following parameters:
- “Show all matching for primers alignment” - checked by default, the software shows the result including all matching of stable binding primer to the target. Combinations of primers not always able to produce the PCR products under the current assay conditions, however the user can check the stability of primer binding sites, orientation and coordinates in the target.
- “Show alignment only for matching primers for PCR product” - in previous option all primer binding sites were represented, while in this case the analysis of primer and target alignment will be shown only for matching primers.
- “Show only amplicons lengths” – this option allows the user to collect only amplicons without analysis of primer and target alignment. This option is recommended to use for In silico PCR of whole genome, including all chromosomes analysis with highly abandoned repeated sequences (In silico PCR for techniques based on repeats: SINE-PCR, IRAP, REMAP, ISSR or RAPD).
The main search criteria for primer binding sites, user can select into the TAB of “Pre-designed Searching Options” that contains:
- “Default criteria searching” - K-mers = 9 with maximum single mismatch at 3' termini (single mismatch within K-mers) and at least 15 bases complement for primer longer 14 nucleotides.
- “Strong criteria searching” - K-mers = 12, single mismatch at 3' termini and at least 15 bases complement for primer longer 14 nucleotides.
- “Sensitive criteria searching” - K-mers = 7 with maximum two mismatch at 3' termini (single mismatch within K-mers) and at least 15 bases complement for primer longer 14 nucleotides.
- “Degenerated sequence”- K-mers = 7 with maximum three mismatch at 3' termini and at least 15 bases complement for primer longer 14 nucleotides.
The “Default criteria searching” is recommended to use to search primer binding sites It provides the most effective and fast search and minimises the false positive results. This criterion is also recommended when user uses degenerated target DNA or/and primers. If necessary the fast searching of primer binding sites with minimum of mismatches at 3' termini, this case user can apply “Strong criteria searching”. In rare cases, when primer sequences or target DNA are degenerate and using of strong criteria of searching did not reveal primer binding sites, it is highly recommended to use the “Sensitive criteria searching” and “Degenerated sequence”. In some cases, user can use the option of “Probe search” or “Linked (Associated) search”. “Probe search” – help user to execute searching of binding sites not only for primers but also for probes (TaqMan, Molecular Beacon, microarrays etc.).
Default value of K-mers: this analysis is set equal to 9 with maximum single mismatch within K-mer. the length of K-mers will be decreased to length of probe if length of probe is shorter than 9 and longer than 3. This option is recommended when primer binding sites are not found or complementarity is expected only for part of a sequence, example in “Molecular Beacon” (both termini have not complementary regions to the target). “Linked (Associated) search” – programmable, advanced sequence searching, when binding sites for primers or probes are searched within determined distance.
Once in silico PCR analysis is completed, the result will be shown in the third Tab Result text editors: In silico PCR Results of the tested targets, where the target is either genomic DNA or sequences whether any targets have been found from the designated genome or sequences. The results will indicate the specificity of the primers (locations, including target position, similarity and Tm). The results will summarise data in primer pairs in relation to the PCR template. Moreover, detailed information on each primer pair, its length and the annealing temperature (Ta) will be provided.
A description line of a primer begins with “In silico Primer(s) search for:” followed by target name and FASTA description of target genome sequence. The description line of a template begins with a “>” sign followed by its identification. In a query primer begins without a “>” sign followed by its identification and including their original sequence. It will show target-specific primers if found, the actual targets will be listed along with detailed alignments between primers and targets.
Based on the tasks, different types of report can be obtained. Individual reports of the query primers include representation of their alignments with the target sequence. The actual targets fragments will be listed along with detailed alignments with linked query sequences and detailed information on each query sequence including locations on target position and its similarity.
Alignment of query primer to their target template will be shown along with their starting and ending coordinates. Nucleotides on the template that match perfectly with the aligned query are embodied by a vertical bar and those mismatched nucleotides are given as a colon (at least similarity 60%) or by a space.
The products are grouped by the target template they are found in. One or multiple products can be found with products size and annealing temperature (Ta) including original primers sequences in FASTA format and including the target position and orientation.
The software allows the user to get information on all potential primer binding sites in DNA target and PCR template, a similar approach can be redundant for the whole genome analysis. In this case, the summary of each primer pair and the length of amplicons will be enough. The user can select or choose a simple report that will contain information only on a PCR size product for each chromosome. Alternatively, the user can select data on only those primers and binding sites that amplify a desired fragment. This is particularly important in PCR performed with genomic DNA as a template.
Linked (Associated) searching
In silico PCR is an example of sequence similarity searching, in which primer sequences are located at a certain distance from each other and are oriented towards each other. A more general method called linked (associated, programmed) searching allows advanced searching of primer-template binding sites in a variety of scenarios, including that of in silico PCR. In linked searching, the search criteria are based only on the distance between annealing sites without taking into account primer orientation and query sequences can be as short as 4 nt.
Another task where linked searching is useful is analysis of two or more nested nucleotide sequences. In the classical in silico PCR, the necessary condition for amplicon search is existence of two primer-binding sites on complementary DNA strands, which are located at a certain distance and orientation relative to each other. In LAMP, the strand displacement-type of DNA amplification, template nucleic acid strands are mixed with three or four nested primer pairs. Linked searching allows the user to analyse primer-template matching during LAMP and predict whether existing primer pairs can amplify a newly discovered sequence variant. Linked searching can be used to quickly determine if the designed LAMP primers can amplify related, consensus or repeated sequences. Such analysis cannot be easily performed with conventional virtual PCR software.
In silico PCR against whole genome(s) or a list of chromosomes
 User must specify a directory by click on any file within a directory.
The program will be sequentially read and analyze each file individually and test primer pairs or the primers (probes) list. Files can contain one or more FASTA sequence with standard IUB/IUPAC nucleic acid codes characters. This software allows simultaneously testing single primer or list of the individual primer or probe with any length thorough multiplex target sequences.
User must specify a directory by click on any file within a directory.
The program will be sequentially read and analyze each file individually and test primer pairs or the primers (probes) list. Files can contain one or more FASTA sequence with standard IUB/IUPAC nucleic acid codes characters. This software allows simultaneously testing single primer or list of the individual primer or probe with any length thorough multiplex target sequences.
Oligo test
Individual oligo are evaluated, it calculate primer Tm’s using default or other formulae for normal and degenerate nucleotide combinations, CG content, extinction coefficient, unit conversion (nmol per OD), mass (µg per OD), molecular weight, linguistic complexity, and primer PCR efficiency. Oligo is analysed for intra- and inter-molecular interactions to form dimers with for standard and degenerate oligonucleotides including LNA and other modifications (U=Uracil; I=Inosine; and LNA: dA=E, dC=F, dG=J, dT=L).
Users can select either DNA or RNA primers with normal or degenerate oligonucleotides or which can be modified with different labels (for example inosine, uridine or fluorescent dyes). Tools allow the choice of other nearest neighbour thermodynamic parameters or simple non thermodynamic Tm calculation formulae. For example, for non-thermodynamic Tm calculation of oligonucleotides, we suggest using the simple formulae, for shorter 15 bases:
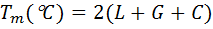
for longer primers, von Ahsen et al. (2001) formulae:
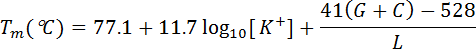 ,
,
where G+C - the number of G's and C's and L - primer length.
Program perform analyses on-type, which allow users to see the results immediately on screen. They can also calculate the volume of solvent require to attain a specific concentration from the known mass (mg), OD or moles of dry oligonucleotide.

Random DNA generation
Using TAB editor, you can select the length (max 10,000 nt) for generation random DNA sequence with “Generate Random DNA”.

Searching for similar sequences (or primers)
FastPCR software allows instant searches for similarities between short or long DNA sequences by simultaneous matching of multiple motifs and can be a useful tool for the identification of distant relationships among DNA sequences and primers.
